Category Archives: drugs
Structure-based drug design updates for SARS-CoV-2
I will be posting here information that is published regarding available crystal structures from SARS-CoV-2 and any other information relevant to structure-based drug design against the virus. Key proteins and their roles in viral infection can be found here. I hope you find this informative and useful for your research.
25 March 2020: 68 crystal structures of SARS-CoV-2 protease, with various bound fragments identified using PanDDA analysis are listed here by EBI.
25 March 2020: The Diamond Light Source (UK) has been able to solve a new structure of the SARS-CoV-2 main protease (MPro) at high resolution (1.25 Å, PDB ID: 6YB7), and subsequently complete a large XChem crystallographic fragment screen against it (detailed here). Data have been deposited with the PDB, and are made available immediately to the world on this page; additional work is ongoing, and updates will be continually posted in their website. On Tuesday March 17th they publicly released results from the full 1500-crystal experiment that yielded 58 non-covalent and covalent active-site fragments. Following data reprocessing and further analysis, an additional 13 structures were released on March 24th taking the final total to 66 active site fragments, 44 of which were covalently bound (full timeline here, download page here). This was an exceptionally large screen, and yielded an exceptionally rich readout, with vast opportunities for fragment growing and merging.
25 March 2020: Blog post, with code by Patrick Walters in Practical Cheminformatics “Building on the Fragments From the Diamond/XChem SARS-CoV-2 Main Protease (MPro) Fragment Screen (Part I)” . Share schemoinformatics techniques and the code used when working with the results of fragment screens.
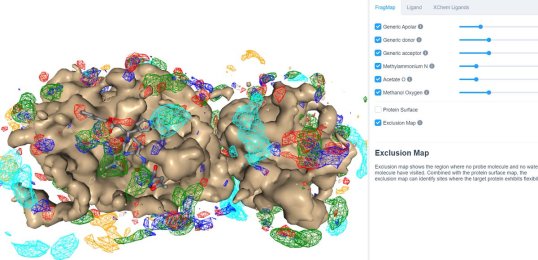
25 March 2020: Visualize SILCS FragMaps for the SARS-CoV-2 main protease (PDB ID 6LU7) through the SILCS demo web viewer, and check out the 6LU7 crystallographic ligand and the XChem fragment screen results from the Diamond Light Source.
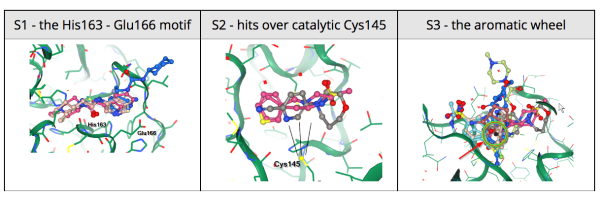
24 March 2020: Results of Diamond Light Source fragment screen:There were 68 hits of high interest – data and extensive details are here, and some interactive views here:
- 22 non-covalent hits in the active site
- 44 covalent hits in the active site
- 2 hits in the dimer interface, one in a calculated hotspot
Based on these data, the team invites chemists around the world to design new compounds or present existing compounds that could bind to the protease and submit them here.Structures submitted are prioritized by factors such as ease of synthesis and potential toxicity and the compounds selected will be synthesized and evaluated for binding to the SARS-CoV-2 protease.
 Results from the XChem fragment screen of the Diamond Light Source can be found here.
Results from the XChem fragment screen of the Diamond Light Source can be found here.
20 March 2020: The crystal structure of SARS-CoV-2 main protease is published complexed with an α-ketoamide inhibitor (PDB ID: 6Y7M). The main viral protease (Mpro, also called 3CLpro) is one of the best characterized drug targets among coronaviruses. 3CLpro protease is required for the virus but lacks human homologous proteins and thus inhibitors of this protease are less likely to bind to a human protease. Research teams in Germany have been able to crystallize the protease and have used this structure to optimize an existing α-ketoamide inhibitor developed to combat other diseases.
 SARS-CoV-2 coronavirus protease dimer bound to an α-ketoamide inhibitor (yellow). The image is reproduced from C&EN news.
SARS-CoV-2 coronavirus protease dimer bound to an α-ketoamide inhibitor (yellow). The image is reproduced from C&EN news.
20 March 2020: SwissProt has modeled the full SARS-CoV-2 proteome based on the NCBI reference sequence NC_045512 which is identical to GenBank entry MN908947, and annotations from UniProt. All data is deposited here.
20 March 2020: Check out refined coordinates of existing experimental SARS-CoV-2 structures using ISOLDE by Tristan Croll (Cambridge University). Modeled coordinates are deposited here.
20 March 2020: Prediction of 10 models for SARS-CoV-2 proteins from the Feig lab.
17 March 2020: The first vaccine clinical trial against SARS-CoV-2 starts in Seattle by Moderna in collaboration with NIAID. The experimental vaccine contains synthetic m-RNA clones that encode glycoprotein S. Once human cells recognize the genetic material of the virus, it is hoped that they will produce antibodies that inactivate the viral glycoprotein S. This strategy is different from the Influenza vaccine, where the viral surface protein itself, hemagglutinin, is introduced into the body and the human immune system produces antibodies against that protein.
5 March 2020: Check out computational predictions of protein structures associated with COVID-19 by DeepMind using AlphaFold, their recently published deep learning system, focuses on predicting protein structure accurately when no structures of similar proteins are available, called “free modelling”.
3 March 2020: Researchers at the Structural Genomic Infectious Diseases Center resolve the crystal structure of SARS-CoV-2 Nsp15 / NendoU endoribonuclease in high resolution. (PDB IDs: 6W01, 6VWW).
20 February 2020: A third research team also published the structure of the SARS-CoV-2 glycoprotein S of SARS-CoV-2 in two conformations using cryo-electron microscopy. (PDB IDs:6VXX, 6VYB)
19 February 2020: A research team from China presents cryo-EM structures of the SARS-CoV-2 glycoprotein S bound to the human ACE2 receptor. The structure shows the full-length human ACE2, in the presence of a neutral amino acid transporter B0AT1, with or without the receptor binding domain (RBD) of the surface spike glycoprotein (S protein) of SARS-CoV-2, both at an overall resolution of 2.9 Å, with a local resolution of 3.5 Å at the ACE2-RBD interface (PDB IDs: 6M18, 6M1D, 6M17).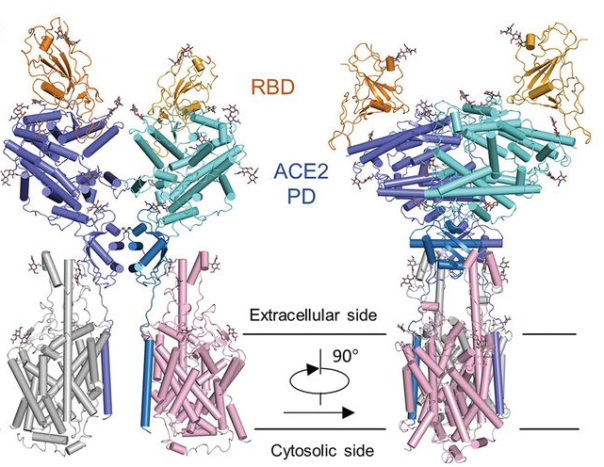 Overall structure of the RBD-ACE2-B0AT1 complex. The complex is colored by subunit, with the ACE2 protease (PD) region depicted in cyan and the Collectrin-like region (CLD) of ACE2 in blue. The glycosylated parts of the human ACE2 receptor are illustrated in strick representation. The RBD subunit of the structure of glycoprotein S that binds to ACE2 is depicted in yellow. The figure has been adapted from the original publication.
Overall structure of the RBD-ACE2-B0AT1 complex. The complex is colored by subunit, with the ACE2 protease (PD) region depicted in cyan and the Collectrin-like region (CLD) of ACE2 in blue. The glycosylated parts of the human ACE2 receptor are illustrated in strick representation. The RBD subunit of the structure of glycoprotein S that binds to ACE2 is depicted in yellow. The figure has been adapted from the original publication.
15 February 2020: The first cryo-EM structure of the viral spike protein (glycoprotein S) is published at a resolution of 3.5 Å (PDB code: 6VSB). Based on the SARS-CoV-2 genome sequence shared by Chinese researchers, they managed to prepare a purified sample of the spike protein and to determine its structure using single particle cryo-EM in less than two weeks. Using surface plasmon resonance they also measured that SARS-CoV-2 glycoprotein S binds 10-20 times more to the human cell ACE2 receptor than the 2002 SARS-CoV virus glycoprotein S. 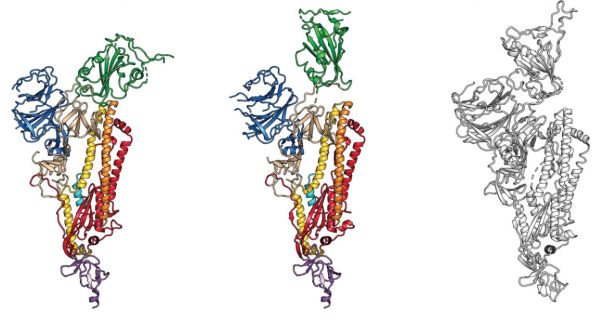 The structure of the glycoprotein S of SARS-CoV-2, as identified by cryo-electron microscopy. Green binds to the binding site that binds to human cells. In the middle image, the green binding domain of glycoprotein S is positioned for binding to the human enzyme ACE2. On the right is the SARS-CoV glycoprotein S for comparison. The figure has been adapted from the original publication.
The structure of the glycoprotein S of SARS-CoV-2, as identified by cryo-electron microscopy. Green binds to the binding site that binds to human cells. In the middle image, the green binding domain of glycoprotein S is positioned for binding to the human enzyme ACE2. On the right is the SARS-CoV glycoprotein S for comparison. The figure has been adapted from the original publication.
5 February 2020: The crystal structure of the SARS-CoV-2 main protease is released in the PDB (PDB ID: 6LU7).
3 February 2020: The complete viral genome of the new coronavirus was analyzed by scientists in China.
30 January 2020: The World Health Organization has named the new virus SARS-CoV-2 and the disease caused by the new virus, COVID-19, and the outbreak was declared a Public Health Emergency of International Concern.
5 January 2020: The source of this infection was quickly identified as a new coronavirus that resembles those that caused SARS-CoV outbreaks in 2002-2004 and MERS-CoV in 2012, but is not the same virus.
31 December 2019: A pneumonia of unknown cause detected in Wuhan, China was first reported to the WHO Country Office in China on 31 December 2019.
Summer of High Performance Computing
Summer of High Performance computing (HPC) is a PRACE programme that offers summer placements at HPC centres across Europe. This year 21 top applicants from across Europe were selected to participate and I was lucky to host two excellent students, Samanta and Juan. They spent two months working on projects in my lab related to HPC.
Samanta worked on understanding the dynamics of the protein human Thymidine Kinase 1 (hTK1), which is crucial for DNA biosynthesis, using Molecular Dynamics simulations. She delivered the project and created an excellent video, I hope you enjoy it!
Juan worked on a computer-aided drug design project. One of the initial steps in drug discovery projects is the virtual screening of small molecules against a set of targets to predict their affinity. Juan created a Python tool has been added to our existing ChemBioServer website that filters compounds so as to be specific for a given set of proteins. He successfully completed the projected and brought new functionality to our server. Enjoy his explanatory video.
Free Virtual Symposium: Advances in Drug Discovery and Development
On September 24, 2014, a world-class group of scientists from biotech, pharma and academia will present the latest research findings and new technologies that are driving the design, discovery and development of new drugs – from small molecules and monoclonals to RNA therapeutics and gene-editing technologies.
 The event is free and organized by the American Chemical Society (ACS) and ACS’s magazine C&EN news.
The event is free and organized by the American Chemical Society (ACS) and ACS’s magazine C&EN news.
To register for free please follow this link. Even if you can’t make the live lectures or the time zone is not convenient for you, all presentations will be archived and available on demand for three months.
You will even have the chance to “Walk the floor” in a virtual exhibition hall, participate in panel discussions and Q&A sessions and do live-chat with peers and vendors
Some of the talks that I find interesting:
Accurate Prediction of Ligand Binding Free Energies – MARK MURCKO, PhD (Disruptive Biomedical LLC)
Design of Protein Structures, Functions and Assemblies – DAVID BAKER, PhD (HHMI/Univ of Washington)
Site-directed ligand discovery for cryptic sites – MICHELLE ARKIN PhD (UCSF)
The Promises and Challenges of Next-Generation Therapeutics – GREG VERDINE, PhD (Warp Drive Bio)
Structure based Drug Design: G-protein coupled receptors using StaR tech – FIONA MARSHALL (Heptares)
Drug Repositioning in the Era of Personalized Medicine – CRAIG WEBB, PhD (NuMedii)
Panel Discussion:The Business of Biotech – LUKE TIMMERMAN, MATT HERPER, MANDY JACKSON
Exploiting cell death for drug discovery – BRENT STOCKWELL, PhD (HHMI/Columbia University)
The VCNDD: CNS Drug Discovery in Academia – CRAIG W. LINDSLEY, PhD (Vanderbilt Center – VCNDD)
Targeting Lysine Acetylation in Cancer – JAY BRADNER, MD (Dana Farber Cancer Institute)
How are drugs designed?
How are drugs designed? Come on Friday 2nd May at 18:00 at the Athens Science Festival, Technopolis Gkazi to listen to my talk! See the detailed program of the festival here.
 This talk will address how drugs are designed to combat a disease – from the discovery of the cause of the disease (e.g. a mutant protein), specialized techniques for the design of small chemical molecules (that are drugs), to clinical trials. We will also discuss advances and progresses in individualized treatment (also known as personalized medicine), i.e. how DNA testing could help each patient receive medication specifically tailored for them.
This talk will address how drugs are designed to combat a disease – from the discovery of the cause of the disease (e.g. a mutant protein), specialized techniques for the design of small chemical molecules (that are drugs), to clinical trials. We will also discuss advances and progresses in individualized treatment (also known as personalized medicine), i.e. how DNA testing could help each patient receive medication specifically tailored for them.
Πώς σχεδιάζονται τα φάρμακα; Ελάτε την Παρασκευή 2 Μαϊου στις 18:00 στο Athens Science Festival, Τεχνοπολις Γκάζι, να ακούσετε την ομιλία μου.
Η ομιλία έχει θέμα το πώς επιτελείται ο σχεδιασμός φαρμάκων για μία ασθένεια – από την ανακάλυψη του αιτίου που προκαλεί την ασθένεια (π.χ. μια μεταλλαγμένη πρωτεΐνη), εξειδικευμένες τεχνικές για το σχεδιασμό μικρών χημικών μορίων που αποτελούν τα φάρμακα, μεχρι και τις κλινικές δοκιμές. Επίσης θα συζητήσουμε τις προοπτικές και εξελίξεις στην εξατομικευμένη θεραπεία, το πώς δηλαδή με εξέταση DNA μπορεί ο κάθε ασθενής να λαμβάνει ένα φάρμακο που είναι ειδικά κατάλληλο για εκείνον.
Περισσότερες πληροφορίες και μία μικρή περίληψη για το θέμα θα βρείτε σε πρόσφατο άρθρο στην ιστοσελίδα της διοργάνωσης.


A Day in the Life of a Computational Chemist
As you know if you have been following this blog, I have always been fascinated by how the world around us works. Why is the sky blue? Why are bubbles in a soft drink spherical? How do we fall in love? What are we really made of?
This inherent curiosity led me to become a scientist. I studied Chemistry but soon enough I realized that being a chemist makes a huge mess or at least I made one in the lab! Fortunately, I then realized that computers exist and they make things much cleaner. I discovered that today it is possible to build chemicals, study reactions, or even make drugs within a desktop computer by performing virtual experiments in a similar way as the typical chemists. This type of chemistry is called “computational chemistry”. So I became a computational chemist. Indeed, I literally live in a virtual reality world, where everything from chemical reactions to drugs, food, materials, cosmetics, electronics, and pr oteins is being modeled and simulated. And you won’t believe it, but, yes, I do have a job!
oteins is being modeled and simulated. And you won’t believe it, but, yes, I do have a job!
I am a group leader at the Biomedical Research Foundation of the Academy of Athens. I specialize in “computer-aided drug design”, so the computer is my Virgil in the world of drugs (to paraphrase the original Nobel Committee tagline). The main activity of my lab is the design of anti-cancer candidate drugs. Recent advances in computer-aided drug design allow us to develop drugs specifically designed for a given protein, shortening the development cycle of new drugs.
Do you want to learn more about what it means to be a computational chemist and how I spend my day? For more details and a video on the life of a computational chemist, please read my full blog post at the Wiley Exchanges site.
Enjoy!
PS. My Doktorvater, Jeremy Smith, was also kind enough to link to this post in his own blog!
Five new candidate drugs are revealed in the ACS New Orleans meeting
The ACS National Spring Meeting took place 7-11 April in New Orleans.
As is traditional for the Medicinal Chemistry Division, structures of candidate drugs get revealed in the “First-Time Disclosures” session.
This year’s entries are:
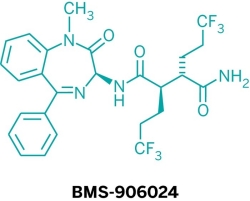


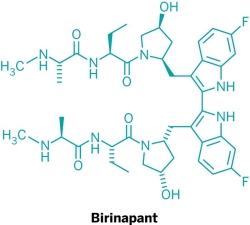

More about how where these drugs discovered:http://cen.acs.org/articles/91/i16/Five-New-Drug-Candidates-Structures.html
Kalydeco: The most important new drug of 2012
Kalydeco, a drug for cystic fibrosis, is the most important new drug of 2012 according to Forbes magazine and was developed by Vertex pharmaceuticals with seed funding from the Cystic Fibrosis Foundation.
Cystic Fibrosis is a genetic disorder that results in scarring (fibrosis) and cyst formation within the pancreas, lungs, liver, and intestines.

Kalydeco’s chemical structure, costing $294,000 per patient per year
Kalydeco, given alone, will only help a few thousand patients the world over. Like other drugs for very rare diseases, its price is very high: $294,000 per patient per year.
Though its chemical structure could be routinely made by a synthetic chemist, it is covered by a patent so it is illegal to make in a lab.
The efforts to cure cystic fibrosis were spearheaded by a discovery from Francis Collins, later famous for heading the Human Genome Project and then the National Institutes of Health, who discovered the gene that, when mutated, causes cystic fibrosis 23 years ago. Kalydeco is the first drug to directly affect the defects caused by these mutations, leading to improvements in patients’ lung function.
