Category Archives: HPC
Structure-based drug design updates for SARS-CoV-2
I will be posting here information that is published regarding available crystal structures from SARS-CoV-2 and any other information relevant to structure-based drug design against the virus. Key proteins and their roles in viral infection can be found here. I hope you find this informative and useful for your research.
25 March 2020: 68 crystal structures of SARS-CoV-2 protease, with various bound fragments identified using PanDDA analysis are listed here by EBI.
25 March 2020: The Diamond Light Source (UK) has been able to solve a new structure of the SARS-CoV-2 main protease (MPro) at high resolution (1.25 Å, PDB ID: 6YB7), and subsequently complete a large XChem crystallographic fragment screen against it (detailed here). Data have been deposited with the PDB, and are made available immediately to the world on this page; additional work is ongoing, and updates will be continually posted in their website. On Tuesday March 17th they publicly released results from the full 1500-crystal experiment that yielded 58 non-covalent and covalent active-site fragments. Following data reprocessing and further analysis, an additional 13 structures were released on March 24th taking the final total to 66 active site fragments, 44 of which were covalently bound (full timeline here, download page here). This was an exceptionally large screen, and yielded an exceptionally rich readout, with vast opportunities for fragment growing and merging.
25 March 2020: Blog post, with code by Patrick Walters in Practical Cheminformatics “Building on the Fragments From the Diamond/XChem SARS-CoV-2 Main Protease (MPro) Fragment Screen (Part I)” . Share schemoinformatics techniques and the code used when working with the results of fragment screens.
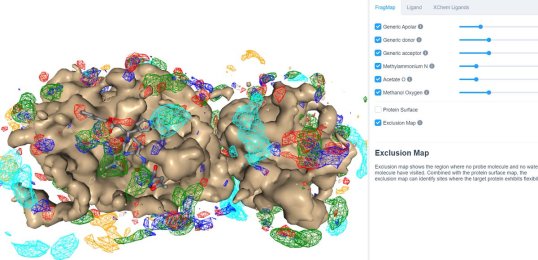
25 March 2020: Visualize SILCS FragMaps for the SARS-CoV-2 main protease (PDB ID 6LU7) through the SILCS demo web viewer, and check out the 6LU7 crystallographic ligand and the XChem fragment screen results from the Diamond Light Source.
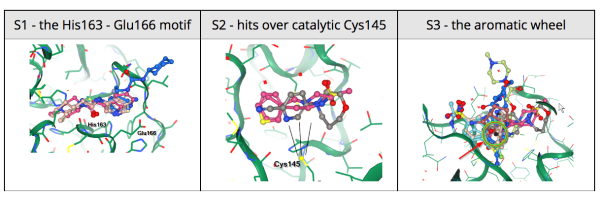
24 March 2020: Results of Diamond Light Source fragment screen:There were 68 hits of high interest – data and extensive details are here, and some interactive views here:
- 22 non-covalent hits in the active site
- 44 covalent hits in the active site
- 2 hits in the dimer interface, one in a calculated hotspot
Based on these data, the team invites chemists around the world to design new compounds or present existing compounds that could bind to the protease and submit them here.Structures submitted are prioritized by factors such as ease of synthesis and potential toxicity and the compounds selected will be synthesized and evaluated for binding to the SARS-CoV-2 protease.
 Results from the XChem fragment screen of the Diamond Light Source can be found here.
Results from the XChem fragment screen of the Diamond Light Source can be found here.
20 March 2020: The crystal structure of SARS-CoV-2 main protease is published complexed with an α-ketoamide inhibitor (PDB ID: 6Y7M). The main viral protease (Mpro, also called 3CLpro) is one of the best characterized drug targets among coronaviruses. 3CLpro protease is required for the virus but lacks human homologous proteins and thus inhibitors of this protease are less likely to bind to a human protease. Research teams in Germany have been able to crystallize the protease and have used this structure to optimize an existing α-ketoamide inhibitor developed to combat other diseases.
 SARS-CoV-2 coronavirus protease dimer bound to an α-ketoamide inhibitor (yellow). The image is reproduced from C&EN news.
SARS-CoV-2 coronavirus protease dimer bound to an α-ketoamide inhibitor (yellow). The image is reproduced from C&EN news.
20 March 2020: SwissProt has modeled the full SARS-CoV-2 proteome based on the NCBI reference sequence NC_045512 which is identical to GenBank entry MN908947, and annotations from UniProt. All data is deposited here.
20 March 2020: Check out refined coordinates of existing experimental SARS-CoV-2 structures using ISOLDE by Tristan Croll (Cambridge University). Modeled coordinates are deposited here.
20 March 2020: Prediction of 10 models for SARS-CoV-2 proteins from the Feig lab.
17 March 2020: The first vaccine clinical trial against SARS-CoV-2 starts in Seattle by Moderna in collaboration with NIAID. The experimental vaccine contains synthetic m-RNA clones that encode glycoprotein S. Once human cells recognize the genetic material of the virus, it is hoped that they will produce antibodies that inactivate the viral glycoprotein S. This strategy is different from the Influenza vaccine, where the viral surface protein itself, hemagglutinin, is introduced into the body and the human immune system produces antibodies against that protein.
5 March 2020: Check out computational predictions of protein structures associated with COVID-19 by DeepMind using AlphaFold, their recently published deep learning system, focuses on predicting protein structure accurately when no structures of similar proteins are available, called “free modelling”.
3 March 2020: Researchers at the Structural Genomic Infectious Diseases Center resolve the crystal structure of SARS-CoV-2 Nsp15 / NendoU endoribonuclease in high resolution. (PDB IDs: 6W01, 6VWW).
20 February 2020: A third research team also published the structure of the SARS-CoV-2 glycoprotein S of SARS-CoV-2 in two conformations using cryo-electron microscopy. (PDB IDs:6VXX, 6VYB)
19 February 2020: A research team from China presents cryo-EM structures of the SARS-CoV-2 glycoprotein S bound to the human ACE2 receptor. The structure shows the full-length human ACE2, in the presence of a neutral amino acid transporter B0AT1, with or without the receptor binding domain (RBD) of the surface spike glycoprotein (S protein) of SARS-CoV-2, both at an overall resolution of 2.9 Å, with a local resolution of 3.5 Å at the ACE2-RBD interface (PDB IDs: 6M18, 6M1D, 6M17).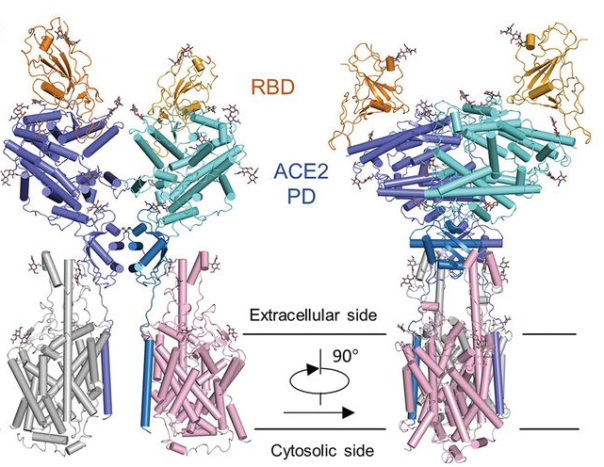 Overall structure of the RBD-ACE2-B0AT1 complex. The complex is colored by subunit, with the ACE2 protease (PD) region depicted in cyan and the Collectrin-like region (CLD) of ACE2 in blue. The glycosylated parts of the human ACE2 receptor are illustrated in strick representation. The RBD subunit of the structure of glycoprotein S that binds to ACE2 is depicted in yellow. The figure has been adapted from the original publication.
Overall structure of the RBD-ACE2-B0AT1 complex. The complex is colored by subunit, with the ACE2 protease (PD) region depicted in cyan and the Collectrin-like region (CLD) of ACE2 in blue. The glycosylated parts of the human ACE2 receptor are illustrated in strick representation. The RBD subunit of the structure of glycoprotein S that binds to ACE2 is depicted in yellow. The figure has been adapted from the original publication.
15 February 2020: The first cryo-EM structure of the viral spike protein (glycoprotein S) is published at a resolution of 3.5 Å (PDB code: 6VSB). Based on the SARS-CoV-2 genome sequence shared by Chinese researchers, they managed to prepare a purified sample of the spike protein and to determine its structure using single particle cryo-EM in less than two weeks. Using surface plasmon resonance they also measured that SARS-CoV-2 glycoprotein S binds 10-20 times more to the human cell ACE2 receptor than the 2002 SARS-CoV virus glycoprotein S. 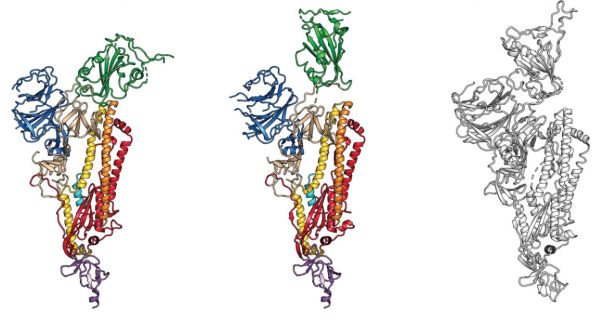 The structure of the glycoprotein S of SARS-CoV-2, as identified by cryo-electron microscopy. Green binds to the binding site that binds to human cells. In the middle image, the green binding domain of glycoprotein S is positioned for binding to the human enzyme ACE2. On the right is the SARS-CoV glycoprotein S for comparison. The figure has been adapted from the original publication.
The structure of the glycoprotein S of SARS-CoV-2, as identified by cryo-electron microscopy. Green binds to the binding site that binds to human cells. In the middle image, the green binding domain of glycoprotein S is positioned for binding to the human enzyme ACE2. On the right is the SARS-CoV glycoprotein S for comparison. The figure has been adapted from the original publication.
5 February 2020: The crystal structure of the SARS-CoV-2 main protease is released in the PDB (PDB ID: 6LU7).
3 February 2020: The complete viral genome of the new coronavirus was analyzed by scientists in China.
30 January 2020: The World Health Organization has named the new virus SARS-CoV-2 and the disease caused by the new virus, COVID-19, and the outbreak was declared a Public Health Emergency of International Concern.
5 January 2020: The source of this infection was quickly identified as a new coronavirus that resembles those that caused SARS-CoV outbreaks in 2002-2004 and MERS-CoV in 2012, but is not the same virus.
31 December 2019: A pneumonia of unknown cause detected in Wuhan, China was first reported to the WHO Country Office in China on 31 December 2019.
Summer of High Performance Computing
Summer of High Performance computing (HPC) is a PRACE programme that offers summer placements at HPC centres across Europe. This year 21 top applicants from across Europe were selected to participate and I was lucky to host two excellent students, Samanta and Juan. They spent two months working on projects in my lab related to HPC.
Samanta worked on understanding the dynamics of the protein human Thymidine Kinase 1 (hTK1), which is crucial for DNA biosynthesis, using Molecular Dynamics simulations. She delivered the project and created an excellent video, I hope you enjoy it!
Juan worked on a computer-aided drug design project. One of the initial steps in drug discovery projects is the virtual screening of small molecules against a set of targets to predict their affinity. Juan created a Python tool has been added to our existing ChemBioServer website that filters compounds so as to be specific for a given set of proteins. He successfully completed the projected and brought new functionality to our server. Enjoy his explanatory video.
